 Homework Assignment #3 and Vector NTI Sessino
for Experiment 5, “Creating new alleles by site-specific mutagenesis and
performing gene knock-ins”
Homework Assignment #3 and Vector NTI Sessino
for Experiment 5, “Creating new alleles by site-specific mutagenesis and
performing gene knock-ins”
You will be performing site-directed mutagenesis of the Aspergillus trtA gene and then “knocking that allele in” to the Aspergillus genome by homologous recombination. Site-directed mutagenesis typically refers to the construction of specifically designed mutant alleles of a gene by changing one or more base pairs in the gene’s sequence. This experimental approach is used for several reasons. For example, it can be used to test the hypothesis that certain amino acids in the protein product of the gene are important for that protein’s function. These amino acids may have been identified as important by studies of homologous proteins from other organisms or have been inferred to be important based on examination of the protein’s 3-D structure. For example, we will test whether a particular aspartate residue (D783) is required for trtA function. This aspartate is present in all reverse transcriptase enzymes. If the Aspergillus trtA gene truly encodes a telomerase reverse transcriptase, then a mutation that removes D783 should inactivate the gene.
Another application of site-directed mutagenesis is to create partial loss of function mutant alleles. This is a particularly useful approach for genes like trtA, where complete loss of function is lethal. Mutants that suffer only partial loss of trtA function should be easier to work with and allow for further analysis of telomerase function in Aspergillus. One convenient class of partial loss of function mutants are temperature sensitive mutants, which appear relatively normal at one temperature but exhibit a mutant phenotype at another temperature. In our experiments, we will create three mutant alleles that are temperature sensitive. At 32 degrees, these mutants will appear to be normal (wild type) but at 43 degrees, they will undergo senescence do to loss of telomerase function.
Each
group will make one mutant allele of trtA in vitro using PCR and then replace
the wild type trtA gene in Aspergillus with the mutant allele. The class will create a total of four
different mutant alleles: L446A, F529A, P539A, and D783A. This nomenclature indicates the amino
acid that is changed by the mutation, the position of that amino acid in 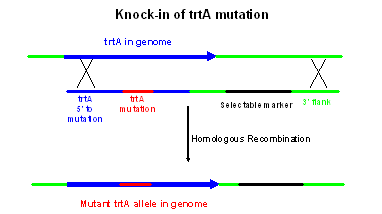 the wild type trtA polypeptide, and the amino acid that is present at
that position in the mutant trtA polypeptide. For example, L446A,
replaces the leucine (L) at position 446 with an alanine
(A). Mutations of the corresponding
amino acids in the yeast telomerase reverse transcriptase have been done and
result in either a temperature sensitive mutation (L446A, F529A, and P539A) or
a null mutation (D783A). I have
provided you with an alignment of telomerase reverse transcriptase proteins
from several organisms as a print out and as a Vector NTI alignment file on
your computer (TERT.apr). Note that
all four amino acids are conserved in at least four of the five proteins in
this alignment.
the wild type trtA polypeptide, and the amino acid that is present at
that position in the mutant trtA polypeptide. For example, L446A,
replaces the leucine (L) at position 446 with an alanine
(A). Mutations of the corresponding
amino acids in the yeast telomerase reverse transcriptase have been done and
result in either a temperature sensitive mutation (L446A, F529A, and P539A) or
a null mutation (D783A). I have
provided you with an alignment of telomerase reverse transcriptase proteins
from several organisms as a print out and as a Vector NTI alignment file on
your computer (TERT.apr). Note that
all four amino acids are conserved in at least four of the five proteins in
this alignment.
Each trtA mutant allele you create will also contain a silent mutation that does not alter the trtA amino acid sequence. The silent mutation will add or remove a restriction site from the trtA gene. Thus, the restriction map of mutant alleles will be different than the wild type allele. This will allow you to confirm that the mutant strains you have created contain the trtA mutation you constructed in vitro by performing a simple restriction digest.
Overview of Experiment 2. The steps for the site-directed mutagenesis project are 1) create a mutant trtA allele using oligonucleotide-mediated site-directed mutagenesis, 2) create a knock-in construct that contains the trtA mutant allele, 3) transform the construct into Aspergillus cells of a strain that primarily uses homologous recombination to insert DNA fragments into its chromosomes, 4) select transformants that are likely to be trtA mutants based on their phenotype, 5) isolate DNA from the selected transformants and use PCR to isolate their trtA gene, and 6) perform restriction enzyme digests to determine whether the trtA gene from these transformants contain the mutation. Instead of analyzing your own mutants, you will be provided with PCR products isolated from all four trtA mutants (L446A, F529A, P539A, and D783A) and you will have to determine which DNA corresponds to which mutant allele.
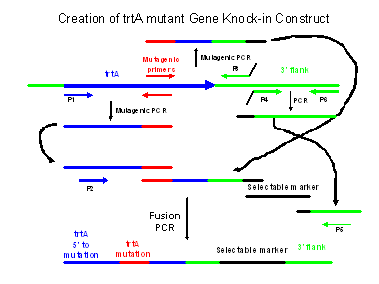
Overview of Steps 1&2: Creating mutant trtA alleles. You will amplify two overlapping portions of trtA using PCR primers that contain the mutant sequence. These primers are referred to as the “mutagenic primers” in the figure to the right. These primers differ from wild type trtA in two ways. First, they change the appropriate trtA codon, creating the amino acid substitution. Second, they change other nearby nucleotides and either create or destroy a restriction site. The overlapping PCR products will be created by using P1 and reverse mutagenic primer in one reaction, and the forward mutagenic primer and P3 in the other. You will use these two trtA fragments, a selectable maker, and the trtA 3’ flanking region, in a fusion PCR reaction with primers P2 and P5. Note that the selectable marker and the 3’ flanking region will be provided to you by the TAs. The result of this PCR reaction will be a fusion product that is your trtA mutant gene knock-in construct.
Overview of Step 3: Transformation of trtA mutant gene knock-in construct into Aspergillus. The trtA mutant gene knock-in construct will be transformed into Aspergillus where it will replace the endogenous trtA gene by homologous recombination, as shown in the figure to the right. This strategy and the strains used are the same as for your gene knock-out in experiment #1, with one exception. The exception is that there is no selection for replacement of the wild type trtA sequence with the mutant sequence you created in vitro. Note that in this figure, I have drawn the crossover events to the right of the selectable marker and to the left of the mutation. These crossovers will indeed replace the wild type trtA sequence with the mutant version. However, transformants will be generated if the crossover on the left occurs anywhere to the left of the selectable marker, including sites that are between the selectable marker and the trtA mutation. Crossover events in that region will yield transformants but they will retain the wild type copy of trtA.
Using Vector NTI to design mutagenic primers. The mutagenic PCR primers must do three things. First, they must be able to anneal to the trtA gene at the site that you want to mutate under the conditions of a PCR reaction. Second, the primers must differ from the trtA coding sequence in a way that results in replacement of one codon for another. Third, they must differ from the trtA sequence in a way that either introduces or destroys a restriction site. The two mutations must be close together, because one mutagenic PCR primer must introduce both mutations.
Let’s look at one of the mutations you will create, D783A, to illustrate these points. To make the D783A mutation, the 783rd codon in trtA must be changed from GAT, which codes for aspartate (D), to either GCT, GCC, GCA, or GCG, which code for alanine (A). The simplest change is a single nucleotide substitution, changing the GAT codon to a GCT codon. The nucleotide change is referred to as an A to C transversion and the corresponding change in the protein encoded by the gene is referred to as an D to A amino acid substitution. Let’s use Vector NTI to look at the wild type trtA and to create a mutant version of trtA corresponding to the L446A mutation. Open the molecule“AN3753.3 trtA region” and select the D783 codon, which is located at 6652 to 6654. You can do this by using the selection box at the bottom right hand side of the window or by scrolling in the text pane and using your cursor to select the sequence. Lets change the A at 6653 to a C using the sequence editor. Select just the A at 6653, then pull down the “edit” menu, choose “new” and “replace sequence…”. Delete the A and type a C in it’s place. Click OK, “keep all”. Save the new molecule as trtA D783AXX using the file menu, and choosing “save as” and remembering to save it in your BIO 510 2008 subset.
Let’s translate exon 6 and to confirm that trtAD783A encodes an A at the site where wild type trtA encodes a D. First, in the trtAD783A molecule, select exon 6 using the graphics pane by clicking on the exon 6 feature. Translate this region by pulling down the “analysis” menu, choosing “translation” then “into sequence pane” and “direct strand”. The “direct strand option always translates the top strand starting at the first nucleotide you have selected, which happens to be the correct reading frame for trtA exon 6. Scroll down the sequence pane to see the codon that starts at position 6652 and note that it is GCT and codes for alanine (A). Now translate exon 6 in the wild type trtA using the same procedure and note that the codon that starts at position 6652 is GAT and codes for aspartate (D). You can confirm that this D is located at position 783 of the trtA protein by comparing the sequence in this region to the sequence of trtA shown on the amino acid alignment. This confirms that your trtAD783A molecule contains a D to A substitution at amino acid 783.
Now lets use Vector NTI to find a way to make a mutation in trtAD783AXX which is close to the A codon at 6652 and which does not change the trtA coding sequence. Vector can search an open reading frame and find all the mutations that can create or destroy restriction sites in that region without introducing mutations that affect the coding sequence. First lest choose three restriction enzymes that will be useful for this purpose and make them the only restriction enzymes displayed. Go to display set up, select restriction enzymes, get rid of all of them except for HindIII, PstI, and XbaI. You may have to add one or more of these enzymes to your display, depending on how you display is set up. Once these are selected, click OK until you done with display set up and have a restriction enzyme folder that contains folders for HindIII, PstI, and XbaI. Note that there are two sites for HindIII, four sites for PstI, and three sites for XbaI. Now select exon 6, which selects proper reading frame for this region of trtA. Now pull down the “analysis” menu, choose “silent mutations” and “direct strand”. This will generate a silent mutations folder that contains all possible simple nucleotide substitutions that will create or destroy sites for these enzymes and are silent with respect to the protein sequence. Scroll down the text pane until mutation #10, which should be a G > T transversion at position 6651, which is supposed to cause a HindIII site to appear. HindIII cuts at AAGCTT and changing the G at 6651 to a T creates a AAGCTT HindIII site.
Lets edit trtAD783AXX to include this mutation so we can use vector to display the HindIII site and to confirm that this mutation is silent with respect to the trtAD783A coding sequence. Select the G at 6651, pull down edit menu, choose “new” and then “replace sequence…”. Delete the G and type in a T. Say OK and “keep all”. Save this molecule by pulling down the file menu, choosing “save as” and naming it trtAD783A XX plus HindIII (save it in the BIO 510 2008 subset). Open the Restriction site folder in the text pane, open the HindIII folder, and notice that there are now three HindIII sites, including one at 6647. This site was not present in trtA. Translate exon 6 as before and note that the translation in this region is the same as that in trtAD783XX.
Your new molecule, trtAD783A plus HindIII XX, is what groups 4 and 8 will make in the D783A site-directed mutagenesis experiment. We will use this molecule to design a mutagenic PCR primer that has the sequence of trtAD783A XX plus HindIII that will work to amplify the wild type trtA sequence using the instructions provided with a site-directed mutagenesis kit marketed by Strategene (called Quick Change). A PDF file version of the manual for this kit is on the class web site FYI. The information from that file that is relevant to mutagenic primer design is included at the end of this document.
Select a region of trtAD783XX plus HindIII starting 20 bp before the T nucleotide you substituted for G through 20 nucleotides after the C nucleotide that you substituted for A. This is from 6631 to 6673. We have selected 20 nucleotides before the first mismatch and 20 nucleotides after the mismatch. This will generally give you a primer that meets the parameters indicated in the QuickChange manual. Lets design mutagenic primers using this sequence and determine whether they meet the required specifications. With 6631 to 6673 selected, use the “add to oligo list” function to add a primer corresponding to the top strand to you list and call it D783AXX. Repeat this process except add an oligo that corresponds to the bottom strand sequence and call it D783AXX complement. Save these oligos to your database. They correspond to the primers group 4 and 8 will use to mutate trtA. All eight mutagenic primers have been provided to you as oligonucleotides in your BIO 510 2008 oligo subset. To double check that the mutagenic oligonucleotides are correct, add them as motifs to the “AN3753 trtA region” and “trtAD783AXX plus HindIII molecules. The match to the trtAD783AXX plus HindIII molecule should be 100% and the match to the wild type molecule should be 95.3%. To calculate the Tm of this primer, you will need to know the number of mismatches. 95.3% identity for a 43 nucleotide oligo is 40.9 or 41 identities and therefore two mismatches.
Some general considerations for designing mutagenic primers using this strategy. Try to design mismatch mutations that are close together, so that your mutagenic primer can have 20 nucleotides of 100% match on either side of the mutations. If this is not possible, then the total length you will select will be greater than 45 nucleotides, which is over the limit according to Strategene. If this is the case, select fewer nucleotides to the left and right of the mismatches and check the Tm of primer. If the shorter primer does not have an appropriate Tm, then make it longer than 45 nucleotides.
Designing mutagenic primers for other exons of trtA. In order to use the “silent mutations” feature of Vector NTI to design silent mutations that alter restriction patterns, you must select the correct open reading frame (ORF). Vector uses the ORF you select to check that its suggested mutations do not change the predicted translation product. Since trtA has 6 exons, determining the correct open reading frame of each exon is a little complicated. To make this easier, let’s create a cDNA-like molecule that only includes the trtA exons, which start at the ATG start codon in exon 1 and end at the TGA stop codon in exon 6. To make this cDNA-like molecule, add exon1, exon2… through exon 6 in order into the fragment goal list. Then construct a linear molecule called trtA cDNA XX and save it in your BIO 510 subset. Note that the trtA cDNAXX molecule retains the exons as annotated features. Selecting the entire molecule and translating it into your sequence pane (analysis>translation>into sequence pane>direct strand) should produce a complete translation of trtA from ATG to TGA. You can also save this translation as a protein molecule in a protein database. Let’s do that by selecting all of trtA cDNA XX and choosing analysis>translation>into new protein>direct strand. Name the protein trtAXX and save it into a new protein database subset, BIO 510 2008. Examine the protein and see that is a 1203 amino acid polypeptide. There should be no * in the protein sequence (* indicates that a non-sense/stop codon was translated). Use this protein sequence file to determine the correct name for any amino acid in trtA. For example, look at position 783 and see that it is a D, which corresponds to the D that will be mutated to an A in the trtAD783A mutant in this lab. You will use this file and the TERT protein alignments to design a site-directed mutagenesis experiment as part of homework 2.
Creating a trtAD783A knock-in construct. The primers you designed for creating the trtAD783A mutant will be used in PCR reactions that generate the 5’ and 3’mutagenic PCR products. These products overlap at the ends corresponding to the mutagenic primer sequences. The two mutagenic PCR products will be used along with the selectable marker and a 3’ flank DNA in a four-way fusion PCR reaction to create the knock-in construct. The selectable marker (Af pyroA Cassette) and the 3’ flank PCR product are already in your BIO 510 2008 database. Let’s make the 5’ and 3’ mutagenic PCR products and then use them to create a knock-in construct that represents what groups 4 and 8 will used to make the trtAD783A mutants.
To create the 5’ and 3’ mutagenic PCR products, open your trtAD783AXX plus HindIII molecule and add the following oligonucleotides as motifs: trtA SD P1, trtA SD P3 no cassette, trtA D783A and trtA D783A complement. When this is done, you should see all the oligos located on trtA in the appropriate locations, matching the sequence with 100% identity. Select from the beginning of the P1 oligo to the end of the trtA D783A complement oligo. To do this using the graphics pane, select the P1 oligo, hold down the shift key, select the D783A complement oligo. Look at the sequence pane to see that the selection is from the beginning of P1 to the beginning of D783A complement. Add this fragment to your fragment goal list. Now select from the beginning of trtA D783A to the end of trtA SD P3 no cassette. Change the region selected to include all the P3 oligo by using the selection box and changing the bottom value in the selection box to 8018. Add this fragment to your fragment goal list. Now open Af pyroA Cassette and add the entire molecule to the fragment goal list. Now open trtA SD 3’ flank and add the entire molecule to the fragment goal list. Now open your fragment goal list and construct the molecule trtA D783A P1 to P6 XX. Use your P1 to P6 molecule to create a P2 to P5 molecule using the same procedure used in experiment 1 for making the knock-out construct.
Restriction analysis to confirm the presence of trtA mutant alleles. In order to confirm that the transformants identified as trtA mutants actually contain the mutant trtA allele you created by PCR, you would need to isolate the trtA gene from the mutants and have it sequenced. Since, we incorporated a second mutation that is detectable by restriction analysis into each of our mutants, we can confirm the presence of the mutant alleles by restriction analysis instead. The first step in this analysis would be to amplify the trtA gene from each mutant using the P1 and P6 primers. You have already created the P1 to P6 PCR product corresponding to the trtA D783A mutant allele, so let’s use that. Open that molecule (trtA D783AXX plus HindIII). Determine the size of restriction fragment generated by digesting with HindIII using analysis>restriction analysis>restriction fragments and then select only HindIII and say OK. Your text pane should now have a restriction fragments folder listing three fragments of 4000 bp, 1129 bp, and 1116 bp. This is the predicted restriction digest pattern for HindIII digestion of the trtA D783A plus HindIII P1 to P6 PCR product. The HindIII site created by the mutation is the site at 2246. If the mutation was not present in your transformant, then the predicted restriction fragment would be two fragments, one of 1116 bp and one of 5129 bp. You should be able to make these same predictions (mutant pattern and wild type pattern) for each of the trtA mutant P1 to P6 products created in this lab.
Information on the mutagenic PCR primers used in this lab.
L446A removes a HindIII site at position 5518 in AN3753 trtA region.
F529A adds a PstI site at position 5824 in AN3753 trtA region
P539A adds a XbaI site at position 5850 in AN3753 trtA region
HOMEWORK 2 DUE by the start of class on Friday, November 20th.
Part 1) Create trtA molecules corresponding to the trtAL446A P1 to P6, trtAF529A P1 to P6 and trtAP539A P1 to P6 mutant alleles you will create in lab. Name them trtAL446AXX P1 to P6, etc.
Part 2) Predict the results of a restriction analysis of P1 to P6 PCR products of all the mutant trtA alleles created in this lab exercise (L446A, F529A, P539A, and D783A).
Part 3) Design mutagenic oligonucleotides that could be used to substitute an alanine (A) for any conserved amino acid in the trtA protein (except do not do L446, F529, P539, or D783). Use the TERT amino acid alignment, the trtA cDNA molecule and the trtA protein molecule to help you with this assignment. The mutagenic oligonucleotides should be useful for mutating the trtA gene (not the cDNA), so avoid choosing codons that are near the splice junctions combining exons 1, 2, 3, 4, 5 and 6. Use the same naming convention used in experiment 2 to name your oligonucleotides, except include your initials in the name.
Email me one molecule archive file and one oligonucleotide archive file that contain your answers to parts 1 and 3. For part 2, include the following in the text of the email:
1) The name of the restriction enzyme that is either created or destroyed by your mutagenic oligo in part 3.
2) Indicate whether that restriction site is created or destroyed.
3) Indicate the predicted sizes of the restriction fragments produced by the analysis in part 2. Be sure to indicate the size of each digest predicted for the wild type and for all four the mutant alleles.